Creutzfeldt–Jakob-kór (CJD): prionbetegség, tünetek és okok
Creutzfeldt–Jakob-kór (CJD): részletes útmutató a prionbetegség tüneteiről, okairól, lefolyásáról és diagnózisáról — ismerje meg a kockázatokat és teendőket.
A Creutzfeldt-Jakob-kór (ejtsd: KROITS-felt YAH-kohb) vagy CJD neurológiai betegség. Degeneratív (idővel rosszabbodik); nem gyógyítható, és mindig halált okoz. A CJD-t néha a "kergemarhakór" (bovine spongiform encephalopathy, vagy BSE) emberi formájának nevezik. A BSE valójában a Creutzfeldt-Jakob-kór egyik ritka típusának egyik oka; a kettő nem ugyanaz a betegség.
A CJD-t egy prionnak nevezett fertőző ágens okozza. A prionok olyan fehérjék, amelyek rosszul vannak összehajtva. A prionok másolatokat készítenek magukról azáltal, hogy a helyesen hajtogatott fehérjéket rosszul hajtogatott formára változtatják. A CJD hatására az agyszövet nagyon gyorsan egészségtelenné válik. Ahogy a betegség elpusztítja az agyat, az agyban lyukak keletkeznek. Az agy szerkezete megváltozik, és olyan lesz, mint egy konyhai szivacs.
Képgaléria
6 Képek




Tünetek
A CJD általában gyorsan progrediáló (romló) tüneteket okoz. A gyakori tünetek:
- gyorsan súlyosbodó demencia (memóriazavar, ítélőképesség és személyiségváltozás);
- mozgászavarok: járászavar, egyensúlyvesztés (ataxia), izomrángások (myoclonus);
- látási problémák, látásromlás vagy hallucinációk;
- beszéd- és nyelészavarok, majd kommunikációs képesség súlyos csökkenése;
- piramidális vagy extrapiramidális tünetek (merevség, lassultság);
- előrehaladott stádiumban akinetikus mutizmus (mozdulatlan, nem beszélő állapot).
A tünetek gyors megjelenése és progressziója (hetek–hónapok alatt) jellemző, ami megkülönbözteti a legtöbb más demenciától, amelyek általában lassabban romlanak.
Okok és típusok
- Sporadikus CJD: a leggyakoribb forma (esetek többsége). Okát nem mindig lehet meghatározni; általában 60 év körüli életkorban kezdődik.
- Genetikai (familialis) CJD: öröklődő mutációk a PRNP génben. A családi formák az összes eset kis részét teszik ki.
- Iatrogén CJD: orvosi beavatkozással átadott esetek (szennyezett idegsebészeti eszközök, dura mater graftok, korábban humán növekedési hormon injekciók); ma ritka a megelőző intézkedések miatt.
- Variant CJD (vCJD): összefüggésbe hozható a BSE-vel (kergemarhakór), jellemzően fiatalabb betegeket érint, gyakran kezdeti pszichiátriai tünetekkel és speciális klinikai/morfológiai jellegzetességekkel.
Hogyan terjed és milyen a lappangási idő?
A prionok nem viselkednek mint a baktériumok vagy vírusok: nem tartalmaznak DNS-t vagy RNS-t, és rendkívül ellenállóak a hagyományos fertőtlenítési módszerekkel szemben. A fertőzés lehetséges mechanizmusai:
- ritkán orvosi átvitellel (iatrogén), például nem megfelelően sterilizált neuroműtéti eszközök vagy szövetátültetések révén;
- variant CJD esetén a szennyezett állati eredetű élelmiszer (BSE-vel szennyezett marhahús) fogyasztása volt a kiváltó mechanizmus a korábbi járványokban;
- családi formában örökletes mutációk adják az okot;
- sporadikus eseteknél a pontos eredet gyakran ismeretlen.
A lappangási idő nagyon változó lehet: néhány évtől több évtizedig terjedhet, különösen iatrogén vagy zoonotikus (állatról emberre) eseteknél.
Diagnózis
A CJD diagnózisa klinikai tünetek, képalkotó és laboratóriumi vizsgálatok összegzésén alapul. Gyakori vizsgálatok:
- neurológiai vizsgálat és a gyorsan progrediáló demencia felismerése;
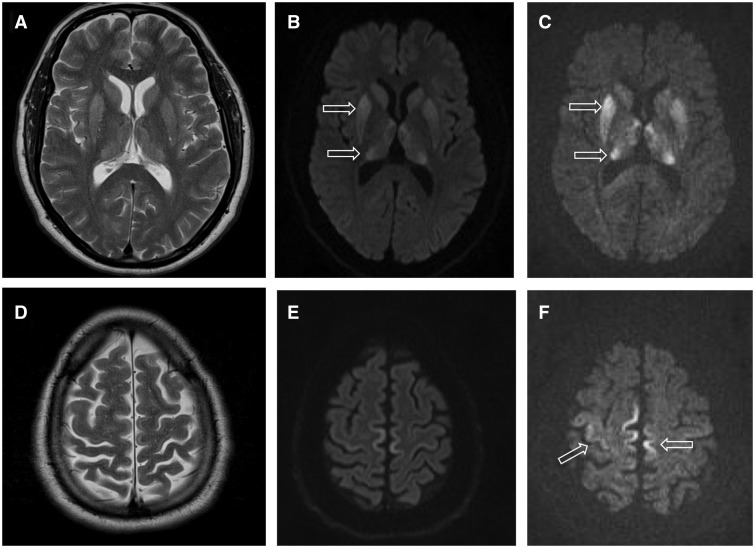
- MRI: diffúziós fázisban jellegzetes elváltozások a kéregben és a bazális ganglionokban;
- EEG: bizonyos típusoknál periodikus éles-hullám komplexek láthatók;
- liquorvizsgálat: 14-3-3 fehérje detektálása korrelálhat a neuronális károsodással (nem teljesen specifikus); a RT-QuIC teszt magas specifitású és érzékeny módszer a prionok kimutatására;
- genetikai vizsgálat PRNP mutációk keresésére, ha öröklődő forma gyanúja áll fenn;
- definitív diagnózis szövettani vizsgálattal (agyszövet biopszia vagy a halál utáni autopszia) érhető el, ahol a spongiform elváltozások és a prionfehérje lerakódások láthatók.
Kezelés és kilátások
Jelenleg nincs gyógyító kezelés a CJD-re. A kezelés tüneti és támogató:
- fájdalom és izomrángások kezelése gyógyszerekkel;
- pszichológiai és ápolási támogatás a betegek és hozzátartozók számára;
- szövődmények (pl. fertőzések, táplálkozási problémák) kezelése.
A betegség általában gyors lefolyású; a sporadikus CJD-ben szenvedők többsége hónapokon belül súlyos állapotba kerül és meghal. A pontos túlélés függ a CJD típusától és az egyéni tényezőktől.
Megelőzés és fertőtlenítés
- orvosi eszközök megfelelő sterilizálása: a prionok ellen speciális, fokozott fertőtlenítési protokollok szükségesek (erősebb vegyszerek — pl. nagy koncentrációjú nátrium-hidroxid vagy hypoklorit — és meghosszabbított, magas hőmérsékletű autoklávozás kombinációja);
- egyszer használatos eszközök alkalmazása nagy kockázatú neuroműtéti beavatkozásoknál;
- vér- és szövetadományozási szabályozások, kockázatos múltú adók kizárása;
- élelmiszer-biztonsági intézkedések a BSE kockázat csökkentésére.
Kutatás
Folyamatban vannak a prionbetegségek megértését és kezelését célzó kutatások: kísérleti gyógyszerek, vakcinák, érzékenyebb diagnosztikai módszerek (pl. RT-QuIC továbbfejlesztése) és a prionfehérje biokémiai viselkedésének tanulmányozása. A prionbiológia megértése fontos lehet más neurodegeneratív betegségek (például Alzheimer-kór) kutatásában is, mert vannak átfedő molekuláris mechanizmusok.
Ha CJD gyanúja merül fel, fontos a gyors neurológiai kivizsgálás és a fertőzés-ellenőrzési intézkedések betartása. A betegek és hozzátartozóik számára palliációs és pszichoszociális támogatás biztonsága kulcsfontosságú.
A CJD típusai és okai
A CJD típusai a következők:
- variáns (vCJD):
A CJD ezen típusát olyan ételek fogyasztása okozhatja, amelyek prionokat tartalmaznak, mint például a BSE-ben ("kergemarhakór") szenvedő tehenek húsa. Ez azonban a CJD nagyon ritka oka.
- sporadikus (sCJD):
Ez a CJD leggyakoribb típusa. A CJD esetek 85%-a sporadikus CJD. Senki sem tudja, hogy mi okozza az sCJD-t; úgy tűnik, hogy véletlenszerűen történik.
- familiáris (fCJD):
A CJD esetek további 15%-ában a legtöbb esetben a CJD családi eredetű. Ez a CJD egy olyan formája, amely családokban fordul elő.
- iatrogén:
A CJD ezen formáját általában olyan orvosi eljárás okozza, amelynek során a személy vérhez vagy szövethez jut egy CJD-ben szenvedő személytől. Például, ha egy személy iatrogén CJD-t kaphat, ha vérátömlesztést vagy szaruhártya-átültetést kap egy CJD-s személytől.
Jelek és tünetek
A CJD első tünete a demencia, amely nagyon gyorsan súlyosbodik.A demencia memóriavesztést, személyiségváltozást és hallucinációkat okoz.
Egyéb gyakori mentális tünetek:
- Szorongás
- Depresszió
- Paranoia
- Kényszerbetegség tünetei
- Pszichózis
A CJD fizikai tünetei gyakran a következők:
- Beszélgetési gondok
- Rángatózó mozgások (myoclonus)
- Egyensúlyzavarok (ataxia)
- Problémák a járással
- Reszketés vagy merevség
- Látási problémák
- Nyelési problémák, amelyek megnehezíthetik vagy lehetetlenné tehetik az étkezést.
- Köhögési zavarok, amelyek tüdőgyulladást okozhatnak.
- Olyan mozgások, amelyeket a beteg nem tud kontrollálni (diszkinézia).
A legtöbb CJD-s ember az első tünetek megjelenését követő hat hónapon belül meghal. Gyakran tüdőgyulladásban halnak meg, amelyet a köhögési zavarok okoznak. A betegek mintegy 15%-a két vagy több évig él. Néhány beteg 4-5 évig élt, többnyire mentális tünetekkel, amíg a betegség súlyosbodik, és több fizikai tünetet okoz. Ha ez megtörténik, az emberek általában egy éven belül meghalnak.
A CJD tüneteit az agy egyre több idegsejtjének elhalása okozza. Amikor a tudósok mikroszkóp alatt megnézik a CJD-s betegek agyszövetét, sok apró lyukat látnak, ahol az idegsejtek egész területei elhaltak.
Diagnózis
Az orvosok CJD-re gyanakodhatnak, ha egy személynek bizonyos tünetei vannak. A demencia például általában lassan romlik. A nagyon gyorsan súlyosbodó demencia szokatlan. Az olyan tünetekkel együtt, mint a rángatózó mozgások, ezek a tünetek utalhatnak a lehetséges CJD-re.
Ezután vizsgálatokkal lehet kimutatni, hogy az illetőnek CJD-e van-e. Ezek a vizsgálatok a következők:
- Elektroenkefalográfia (EEG): Ez a vizsgálat az agy elektromos aktivitását mutatja. Az orvos gyakran képes lesz látni az EEG-ben olyan változásokat, amelyek gyakoriak a CJD-ben szenvedőknél. Az EEG-n látható változások típusa attól függ, hogy a betegnek milyen típusú CJD-je van, és milyen előrehaladott állapotban van a beteg.
- Lumbálpunkció (gerinccsapolás): Ez a vizsgálat lehetővé teszi az agy-gerincvelői folyadék (az agyat és a gerincvelőt körülvevő folyadék) vizsgálatát egy specifikus fehérje ("14-3-3 fehérje") keresésével.
- Az agy MRI-je: A vizsgálat során egy nagyon erős mágnes segítségével képeket készítenek az agyról.
- Biopszia: A biopszia elvégzéséhez a sebész egy tű segítségével egy kis szövetdarabot vesz ki a testből, hogy az orvosok mikroszkóp alatt megnézhessék azt.A vCJD-t a mandulákból vett biopsziával lehet diagnosztizálni. A CJD minden más típusa esetében az agyból vett biopszia az egyetlen módja annak, hogy biztosan megállapítsák, hogy az illetőnek CJD-e van. Mivel azonban az agy biopsziája agykárosodást okozhat, az agyi biopsziát általában nem végzik el, ha más vizsgálatok már kimutatták, hogy a személy valószínűleg CJD-s.

Kezelés
2016-ban még nem létezik olyan kezelés, amely gyógyítja a CJD-t, vagy akár csak lassítja annak hatásait. Számos kísérletet végeznek a kezelések megtalálására.
Ma a CJD egyetlen kezelése olyan gyógyszerek, amelyek a betegség tüneteit kezelik, és segítenek a betegeknek, hogy jobban érezzék magukat. Például a rohamot kapó betegek görcsoldó gyógyszereket kaphatnak. A benzodiazepinek hatására az izomrángások ritkábban fordulhatnak elő.
A betegek dönthetnek úgy is, hogy a rossz tünetek enyhítésére orvosi beavatkozásokat vesznek igénybe. A CJD például olyan mértékű nyelési problémákat okozhat, hogy az illető nem tud enni. Néhány CJD-s ember úgy dönt, hogy tápszondát helyez be, amikor már nem tud enni. Ez egy olyan cső, amely a gyomorba vezet, így speciális folyadékot lehet közvetlenül a gyomorba juttatni, hogy a személy táplálékot kapjon.
Kapcsolódó oldalak
- Prion
- Prion betegség
- Halálos betegség
Kérdések és válaszok
K: Mi az a Creutzfeldt-Jakob-kór?
V: A Creutzfeldt-Jakob-kór (CJD) egy degeneratív, gyógyíthatatlan és mindig halálos kimenetelű idegrendszeri betegség.
K: Van-e gyógymód a CJD-re?
V: Nem, a CJD-re nincs gyógymód.
K: Miért nevezik a CJD-t néha a "kergemarhakór" emberi formájának?
V: A CJD-t néha a "kergemarhakór" emberi formájának nevezik, mert a szarvasmarhák szivacsos agyvelőbántalma (BSE), amely a CJD egyik ritka típusát okozza, általánosan "kergemarhakór" néven ismert.
K: Mi a CJD oka?
V: A CJD-t egy prionnak nevezett fertőző ágens okozza, amely egy olyan fehérje, amely helytelenül hajtogatott, és képes másolatokat készíteni magáról azáltal, hogy a megfelelően hajtogatott fehérjéket rosszul hajtogatottakká változtatja.
K: Mi történik az agyszövetben a CJD esetén?
V: A CJD hatására az agyszövet nagyon gyorsan egészségtelenné válik, ami lyukak kialakulásához vezet az agyban, és az agy textúrájának megváltozásához, amely olyan lesz, mint egy konyhai szivacs.
K: A BSE ugyanaz a betegség, mint a CJD?
V: Nem, a BSE nem ugyanaz a betegség, mint a CJD; valójában a CJD egy ritka típusának egyik oka.
K: Hogyan okozzák a prionok a CJD-t?
V: A prionok azáltal okoznak CJD-t, hogy helytelenül hajtogatódnak és másolatokat készítenek magukról a helyesen hajtogatott fehérjék rovására az agyban. Ez az egészséges agyszövet pusztulását és a betegségre jellemző lyukak kialakulását eredményezi.
Kapcsolódó cikkek
Szerző
AlegsaOnline.com Creutzfeldt–Jakob-kór (CJD): prionbetegség, tünetek és okok Leandro Alegsa
URL: https://hu.alegsaonline.com/art/24152
Források
- cdc.gov : "CJD (Creutzfeldt–Jakob Disease, Classic)"
- ncbi.nlm.nih.gov : "Creutzfeldt–Jakob disease: Transmissible spongiform encephalopathy; vCJD; CJD; Jacob-Creutzfeldt disease"
- bmj.com : "Bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease"
- doi.org : 10.1101/SQB.1996.061.01.052
- pubmed.ncbi.nlm.nih.gov : 9246478
- www3.interscience.wiley.com : "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course"
- doi.org : 10.1111/j.1440-1789.2008.00904.x
- pubmed.ncbi.nlm.nih.gov : 18410280
- who.int : who.int: "Fact sheets no 180: Variant Creutzfeldt-Jakob disease" Feb 2012 ed.
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- doi.org : 10.1056/NEJM198610163151605
- pubmed.ncbi.nlm.nih.gov : 3762620
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- merckmanuals.com : "Creutzfeldt–Jakob Disease (CJD)"
- ncbi.nlm.nih.gov : "Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy"