Progeroid szindrómák — meghatározás, okok és fő típusok (HGPS, Werner)
Ismerje meg a progeroid szindrómák meghatározását, okait és fő típusait (HGPS, Werner): genetika, tünetek, diagnózis és kutatási kilátások.
A progeroid szindrómák (PS) olyan, öröklött betegségek csoportját jelentik, amelyekben a betegek szerveiben és szöveteiben az öregedés bizonyos jellegzetességei felgyorsult formában jelennek meg. Ezek a rendellenességek legtöbbször monogenetikus eredetűek: egyetlen gén mutációjából adódnak, és sok esetben a hibák vagy a DNS-javító mechanizmusban, vagy a magvázhoz kapcsolódó, lamin A/C típusú fehérjék működésében keresendők.
A „progeroid” szó jelentése „öregségre emlékeztető”: ez az elnevezés arra utal, hogy az érintettek bizonyos tekintetben az időskorban megszokott elváltozásokat mutatnak, de nem feltétlenül a természetes öregedés minden aspektusát. Például az Alzheimer-kór vagy a Parkinson-kór jellemzői általában egy-egy szervre koncentrálódnak, míg a progeroid szindrómákban többféle szövet — bőr, kötőszövet, csont, érrendszer, endokrin szervek — is érintett lehet.
A PS-csoporton belül a legjobban ismert és legintenzívebben kutatott formák a Hutchinson–Gilford progeria syndrome (HGPS) és a Werner-szindróma (WS). A HGPS gyermekkorban manifesztálódik és rendkívül gyors fizikai öregedéssel jár; a WS általában fiatal felnőttkorban kezdődik, és a klinikai képe az időskori betegségek korai megjelenését idézi. Emiatt ezek a betegségek fontos modelljei az öregedés, a regeneráció, az őssejtek biológiája és a rák kutatásának.
Képgaléria
3 Képek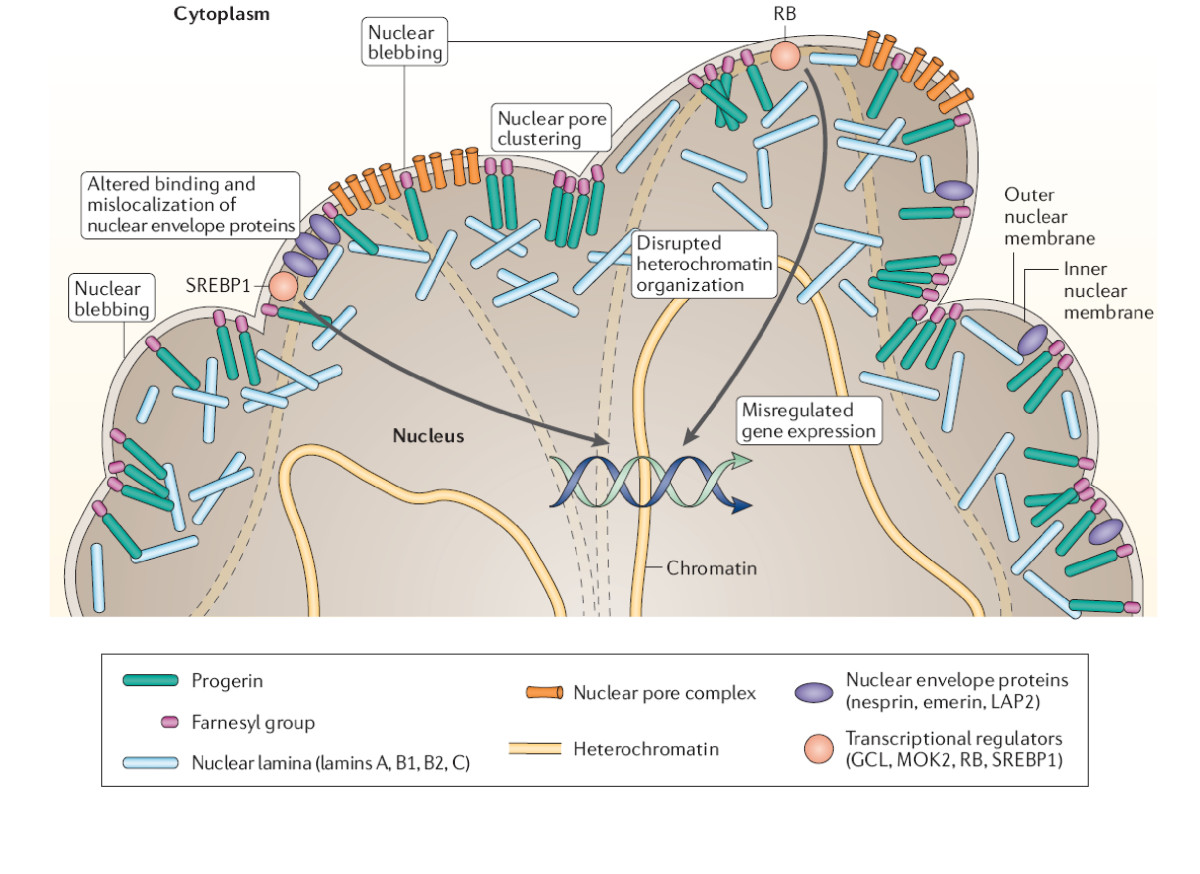

Okok és molekuláris mechanizmusok
- LMNA gén és progerin (HGPS): a HGPS leggyakoribb oka egy szinonim mutáció (például G608G) az LMNA génben, ami hibás hasadást (cryptic splicing) eredményez és egy rövidebb, toxikus lamin A fehérje, az ún. progerin kialakulásához vezet. A progerin megváltoztatja a sejtmag szerkezetét és a kromatin működését, ezáltal sejtinstabilitást és gyors szeneszcenciát vált ki.
- DNS-javítási hibák: több progeroid syndroma oka a DNS-javítást végző gének károsodása. Ilyen például a Werner-szindróma, amelyet a WRN (RECQL2) helicáz gén mutációja okoz; ennek következménye a fokozott genominstabilitás és a korai daganatosodásra való hajlam.
- Más genetikai okok: számos ritkább progeroid forma összefügg többek között a RECQL4 (Rothmund–Thomson), BLM (Bloom-szindróma) génekkel vagy a ZMPSTE24 proteázzal, amelyek szintén a sejtmag, a kromatin vagy a DNS-javító folyamatok működését befolyásolják.
Klinikai jellegzetességek
A tünetek és a súlyosság típusonként változnak, de gyakori jellemzők:
- ritkás, elvékonyodó haj, kopaszodás;
- vékony, ráncos bőr, pigmentációs eltérések;
- alacsony növekedés és általános fejlődési elmaradás (különösen HGPS-ben);
- korai csontvesztés, osteoporózis és izomgyengeség;
- korai ateroszklerózis és szív-ér rendszeri betegségek (ez HGPS-ben gyakori halálok);
- korai cataracta, endokrin eltérések (pl. diabetes), szubfertilitás;
- megnövekedett daganatos kockázat bizonyos típusokban (például Werner-szindrómában).
Diagnózis
A diagnózis általában a jellegzetes klinikai kép és a családi anamnézis alapján merül fel, és megerősítéséhez molekuláris genetikai vizsgálat szükséges. A célzott genetikai tesztelés az érintett gén(ek) mutációinak kimutatására szolgál (pl. LMNA a HGPS-ben, WRN a Werner esetében). Emellett fontos a kardiovaszkuláris státusz, anyagcsere-paraméterek, csontsűrűség és onkológiai szűrések rendszeres nyomon követése.
Kezelés és gondozás
Jelenleg nincs általános gyógyító terápia a legtöbb progeroid szindrómára, így a kezelés elsősorban tüneti és szövődmény-orientált. A gondozás több szakterület összehangolt munkáját igényli:
- kardiológiai és érvizsgálatok rendszeres elvégzése, kockázatcsökkentő intézkedések;
- metabolikus és endokrin gondozás (pl. vércukor, lipidek kezelése);
- ortopédiai és fizioterápiás ellátás a mozgásfenntartásért;
- dermatológiai, szemészeti és fogászati kontroll;
- dohányzás- és kardiovaszkuláris rizikó faktorok szigorú kezelése;
- daganat-szűrések a hajlamosító formákban;
- pszichoszociális támogatás és családi tanácsadás.
Klinikai kutatások és egyedi terápiák is folynak: HGPS-ben például a farnesiltranszferáz-gátlók (pl. lonafarnib) alkalmazása javította bizonyos betegek túlélését és kardiovaszkuláris állapotát; vizsgálják továbbá antiszenz-oligonukleotidokat, géniásítási megközelítéseket és kombinált gyógyszeres terápiákat.
Prognózis és epidemiológia
A prognózis nagyon függ a pontos típustól. A HGPS jellemzően gyermekkorban okoz súlyos érrendszeri betegséget és a korai halálozás fő oka szív- és érrendszeri komplikáció. A Werner-szindrómában a túlélés gyakran 40–50 éves kor körül csökken, de ez egyénenként változó, és a betegek életminősége a gondozás minőségétől függ. A progeroid szindrómák ritkák: a HGPS előfordulása rendkívül alacsony (millió születésre jutó esetek), míg a WS előfordulása bizonyos népességekben, például Japánban, nagyobb lehet.
Genetikai tanácsadás és megelőzés
Mivel sok progeroid szindróma monogenetikus, a genetikai tanácsadás fontos: érintett családokban ajánlott a hordozóság vizsgálata, a reprodukciós opciók megbeszélése, valamint szükség esetén prenatális vagy preimplantációs genetikai diagnosztika. A korai felismerés és a rendszeres, multidiszciplináris gondozás javíthatja a betegek életminőségét és csökkentheti a szövődmények kockázatát.
Kutatás és jövő
A progeroid szindrómák kutatása kettős előnyt nyújt: egyrészt közvetlenül célzott terápiák fejlesztését teszi lehetővé a ritka betegségekben szenvedők számára, másrészt modellekként szolgál az általános öregedési folyamatok megértéséhez. A laboratóriumi modellek, őssejt-alapú vizsgálatok, preklinikai gyógyszerkutatások és génszerkesztési technikák ígéretes irányok, amelyek a közeljövőben tovább bővíthetik a terápiás lehetőségeket.
Összefoglalva: a progeroid szindrómák ritka, de nagyon informatív betegségek, amelyek a genetikai hibák és a sejtszintű öregedési mechanizmusok jobb megértéséhez vezetnek. A Werner-szindróma és a Hutchinson-Gilford-progéria-szindróma (HGPS) a legjobban ismert formák közé tartoznak, és mind diagnosztika, mind gondozás, mind kutatás tekintetében speciális megközelítést igényelnek.
Kérdések és válaszok
K: Mi az a progeroid szindróma (PS)?
V: A progeroid szindrómák egy sor olyan genetikai rendellenesség, ahol az érintett személy gyorsabban öregszik.
K: Mi okozza a PS-t?
V: A legtöbb ismert PS-mutáció vagy a DNS-javító mechanizmusban, vagy a lamin A/C nevű fehérje hibáihoz vezet.
K: Mit jelent a progeroid?
V: A progeroid azt jelenti, hogy öregségre hasonlít.
K: Az Alzheimer-kór és a Parkinson-kór tekinthető progeroid szindrómának?
V: Nem, az Alzheimer- és a Parkinson-kór csak egy szövetet érint, és a progeroid szindróma kifejezést olyan esetekre használják, amikor az érintettek csak az öregedés egyes jellemzőit mutatják, de nem az összeset.
K: Hányféle szövetet érinthet a progeroid szindróma?
V: A progeroid szindrómákban sokféle szövet érintett.
K: Mekkora a PS-szel kapcsolatos rendellenességekben szenvedő egyének élettartama?
V: A PS-szel kapcsolatos rendellenességekben szenvedő egyének élettartama gyakran csökken.
K: Melyek a legszélesebb körben vizsgált progeroid szindrómák, és miért?
V: A legszélesebb körben vizsgált progeroid szindrómák a Werner-szindróma (WS) és a Hutchinson-Gilford-progéria-szindróma (HGPS), mivel ezek hasonlítanak a természetes öregedésre.
Kapcsolódó cikkek
Szerző
AlegsaOnline.com Progeroid szindrómák — meghatározás, okok és fő típusok (HGPS, Werner) Leandro Alegsa
URL: https://hu.alegsaonline.com/art/79367
Források
- doi.org : 10.1093/hmg/ddl214
- pubmed.ncbi.nlm.nih.gov : 16987878