Osteogenesis imperfecta: Törékeny csontok genetikai rendellenessége
Osteogenesis imperfecta — genetikai törékeny csontok: tünetek, típusok, öröklődés és kezelési lehetőségek. Ismerje meg a diagnózist, megelőzést és életminőség-javítást.
Osteogenesis imperfecta genetikai rendellenesség, amelyet gyakran törékeny csontbetegségnek neveznek. A betegség a csontok szerkezetének egyik kulcselemét, a kollagént érinti: a kóros gén a kollagén felépítését és szerkezetét gyengíti, ezáltal csökkentve a csontok teherbírását. A mutációk többnyire a COL1A1 vagy COL1A2 géneket érintik, amelyek a csont és kötőszövet fő komponensét, az I-es típusú kollagént kódolják. Ezt a betegséget először Vrolik azonosította 1854-ben.
Képgaléria
10 Képek







Okok és öröklődés
Az OI legtöbbször autoszomális domináns módon öröklődik, ami azt jelenti, hogy egyik érintett szülő is elegendő a betegség kialakulásához. Ugyanakkor ismertek autoszomális recesszív formák is, valamint újonnan keletkező (de novo) mutációk, amikor a családban korábban nem volt érintett. A mutációkat gyakran a génnel azonosítják molekuláris genetikai vizsgálattal.
Tünetek
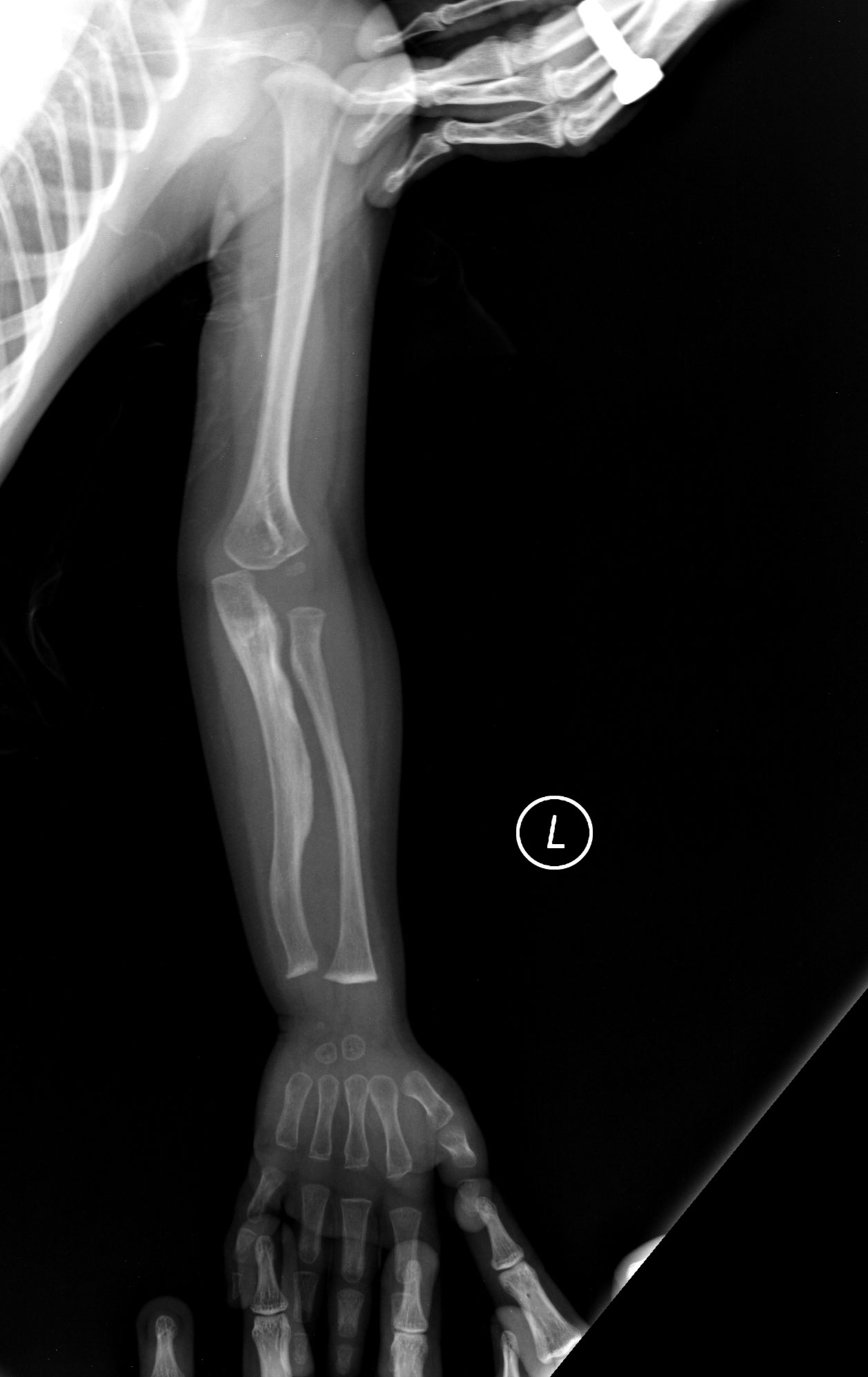
- Gyakori csonttörések, akár kisebb traumára is
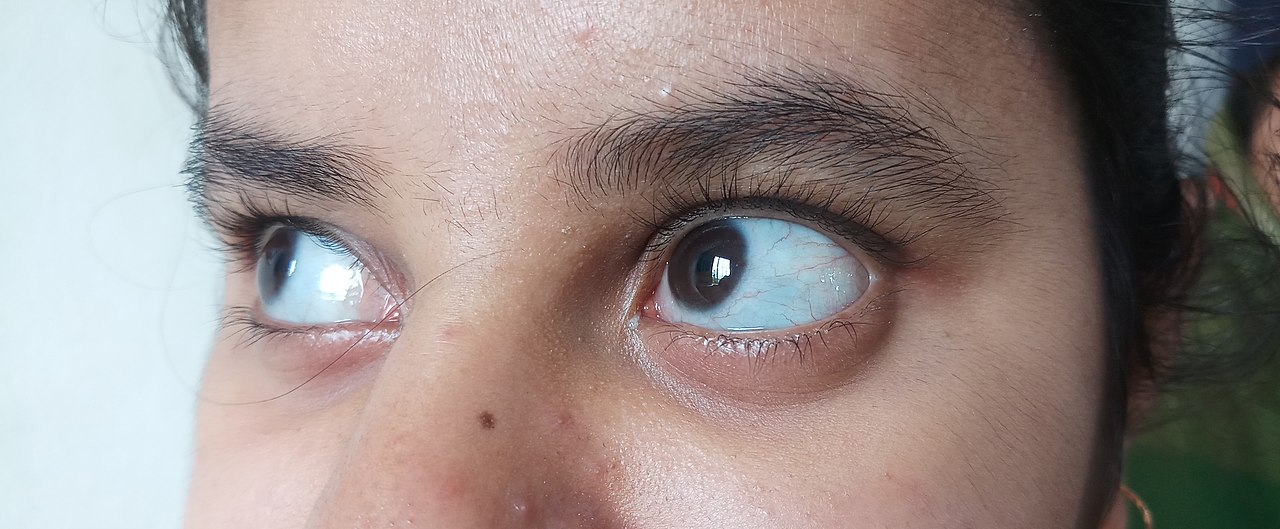
- Kékes, lila vagy szürkés szemfehérje (kék sclera), ami a szem erezettségének és a kötőszövet átlátszóságának következménye
- Dentinogenesis imperfecta (törékeny, elszíneződött fogak)
- Halláscsökkenés vagy hallásvesztés (gyakran felnőttkorra alakul ki)
- Rövid növés, csontdeformitások (pl. görbült csontok, scoliosis)
- Izom–ízületi lazaság és mozgáskorlátozottság súlyosabb esetekben
Típusok (Sillence-osztályozás, főbb csoportok)
- 1. típus (enyhe): leggyakoribb; normál vagy közel normál testmagasság, törékeny csontok, gyakran kék sclera és dentinogenesis.
- 2. típus (perinatálisan halálos): leg súlyosabb forma; gyakran légzési nehézségek és súlyos csontdeformitások miatt a korai életévekben halálozás fordul elő.
- 3. típus (súlyos, progresszív): nagyon gyakoriak a törések a korai gyermekkorban, jelentős csontdeformitások és rövid növés jellemzi; pubertás előtt több tucat vagy akár több száz törés is előfordulhat.
- 4. típus (közepes súlyosságú): tünetei súlyossága a 1. és 3. típus közé esik; változó fokú törékenység és deformitás jellemzi.
Diagnózis
A diagnózis alapja a kórtörténet (gyakori törések, családi anamnézis), fizikai vizsgálat és képalkotó vizsgálatok (röntgen). A genetikai vizsgálat a pontos molekuláris okot képes feltárni, és segít a típus meghatározásában. Bizonyos esetekben kollagénanalízis bőr- vagy kötőszöveti mintából is elvégezhető.
Kezelés és gondozás
Az OI nem gyógyítható, de a tünetek és szövődmények kezelhetők, és a betegek életminősége javítható. A kezelés általában multidiszciplináris:
- Fraktúrák ellátása és megelőző ortopédiai beavatkozások (pl. intramedulláris rúdbeültetés a hosszú csontok stabilizálására)
- Fizikoterápia és gyógytorna a mozgásképesség javítására és az izomerő fenntartására
- Biszfoszfonátok (pl. pamidronát) adása bizonyos gyermekek és felnőttek esetében a csontsűrűség növelésére és a törések csökkentésére
- Fájdalomkezelés és rehabilitáció
- Fogászati ellátás dentinogenesis miatt
- Hallásvizsgálat és szükség esetén hallókészülék
- Genetikai tanácsadás a családok számára, tervezett terhességek esetén
Prognózis és életminőség
A kórlefolyás nagyon változó: a 2. típus általában perinatálisan súlyos kimenetelű, míg az 1., 3. és 4. típusok esetén a várható élettartam és életminőség széles skálán mozog, nagymértékben függ a betegség súlyosságától és a kapott kezeléstől. Sok beteg önálló életvitelre képes, ha megkapja a megfelelő orvosi és rehabilitációs támogatást.
Gyakorlati tanácsok
- Olyan életmód és környezet kialakítása, ami csökkenti a sérülések kockázatát (otthoni biztonság, megfelelő sportok kiválasztása)
- Rendszeres orvosi kontrollok: ortopéd, endokrinológus, genetikus, fogorvos és audiológus bevonásával
- Genetikai tanácsadás a családtervezéshez és a rizikó felméréséhez
Az OI kezelésében és kutatásában folyamatos fejlődés tapasztalható; fontos a korai diagnózis és a személyre szabott, multidiszciplináris ellátás. Ha több információra van szüksége a csontok felépítéséről, lásd: csont.
Tünetek
Az OI kevésbé súlyos tünetei a következők lehetnek:
- könnyen törött csontok
- laza ízületek
- alacsony izomtónus
- kék, lila vagy szürke szín a szem normális esetben fehér részén.
- háromszög alakú arc
- skoliózis kialakulására való hajlam
- törékeny fogak
Az OI-nek számos más súlyos és végzetes tünete is van, beleértve a légzési problémákat és a csontdeformitást.
Demográfiai adatok
Az OI férfiaknál és nőknél egyaránt előfordul, és minden etnikai csoportot érinthet. Az OI már az anyaméhben kialakul, és nem gyógyítható. Általában akkor fedezik fel, amikor valaki sok csontot tör; DNS-vizsgálatot végezhetnek az OI kimutatására. Húszezer születésből 1 esetben fordul elő.
Kérdések és válaszok
K: Mi az osteogenesis imperfecta?
V: Az osteogenesis imperfecta egy genetikai rendellenesség, amelyet általában törékeny csontbetegségnek neveznek. Gyengíti vagy elpusztítja a kollagénrudat, amely a csont szilárdságát biztosítja, és olyan csontokhoz vezet, amelyek nagyobb valószínűséggel törnek el.
K: Hogyan öröklődik az osteogenesis imperfecta?
V: Az osteogenesis imperfecta általában autoszomális domináns betegség, ami azt jelenti, hogy egy személy akkor kaphatja meg, ha csak az egyik szülője rendelkezik a kóros génnel.
K: Ki azonosította először az osteogenesis imperfectát?
V: Vrolik azonosította először az osteogenesis imperfectát 1849-ben.
K: Gyógyítható-e az osteogenesis imperfecta?
V: Sajnos az osteogenesis imperfecta nem gyógyítható.
K: Mi az osteogenesis imperfecta négy típusa?
V: Az osteogenesis imperfecta négy típusa az egyes, a kettes, a hármas és a négyes típus.
K: Milyen tünetei vannak a hármas típusú osteogenesis imperfectának?
V: A hármas típusú osteogenesis imperfectában szenvedő embereknek több mint 100 törésük lehet a pubertáskor előtt. A szemük gyakran lila, kék vagy szürke árnyalatú, és az ebben az esetben érintettek gyakran halláskárosodást is tapasztalnak.
K: Mennyire gyakori a halláskárosodás az osteogenesis imperfecta-ban szenvedő embereknél?
V: Az osteogenesis imperfectában szenvedő emberek 50%-ánál felnőtté válásuk során halláscsökkenés jelentkezik.
Kapcsolódó cikkek
Szerző
AlegsaOnline.com Osteogenesis imperfecta: Törékeny csontok genetikai rendellenessége Leandro Alegsa
URL: https://hu.alegsaonline.com/art/73422
Források
- oif.org : "Osteogenesis Imperfecta Foundation:"
- nlm.nih.gov : "Autosomal dominant: MedlinePlus Medical Encyclopedia"
- oif.org : "Osteogenesis Imperfecta Foundation: Understanding Bone Structure"