Thalassaemia – meghatározás, okai, tünetei és kezelése
Thalassaemia — meghatározás, okai, tünetei és kezelése: érthető, gyakorlatias útmutató öröklődésről, diagnózisról, szövődményekről és modern terápiákról.
A thalassaemia (vagy thalassemia) a vér genetikai rendellenessége, amely a mediterrán térségből származik.
Ezt a betegséget a vörösvértestek gyengülése és pusztulása okozza. Ezt olyan mutáns gének okozzák, amelyek befolyásolják a szervezet hemoglobintermelését. A hemoglobin a vörösvértestekben található fehérje, amely az oxigént szállítja. A thalassaemiás emberek a normálisnál kevesebb hemoglobint és kevesebb keringő vörösvértestet termelnek, ami enyhe vagy súlyos vérszegénységet eredményez.
A thalassaemia jelentős szövődményeket okozhat, többek között tüdőgyulladást, vastúlterhelést, csontdeformitást és szív- és érrendszeri betegségeket. A vörösvértestek ezen örökletes betegsége azonban bizonyos fokú védelmet nyújt a malária ellen, amely gyakori vagy gyakori volt azokban a régiókban, ahol a tulajdonság gyakori. A hordozóknak ez a szelektív túlélési előnye (az úgynevezett heterozigóta előny) felelős azért, hogy a mutáció a populációkban jóval a mutációs arány felett marad. A hordozók heterozigóták a thalassaemia allél tekintetében, ami azt jelenti, hogy a két allél közül csak az egyik mutáns (abnormális). A thalassaemiának számos különböző változata létezik. Mindegyiket a genom egy-egy különböző pozíciójában bekövetkező mutáció okozza.
Ebben a tekintetben a thalassaemia hasonlít egy másik, a hemoglobint érintő genetikai rendellenességhez, a sarlósejtes betegséghez.
A thalassaemiás betegek gyógyítása kompatibilis donoroktól származó csontvelőátültetéssel lehetséges. Ehhez a módszerhez azonban HLA-egyeztetett kompatibilis donorra van szükség.
Képgaléria
8 Képek



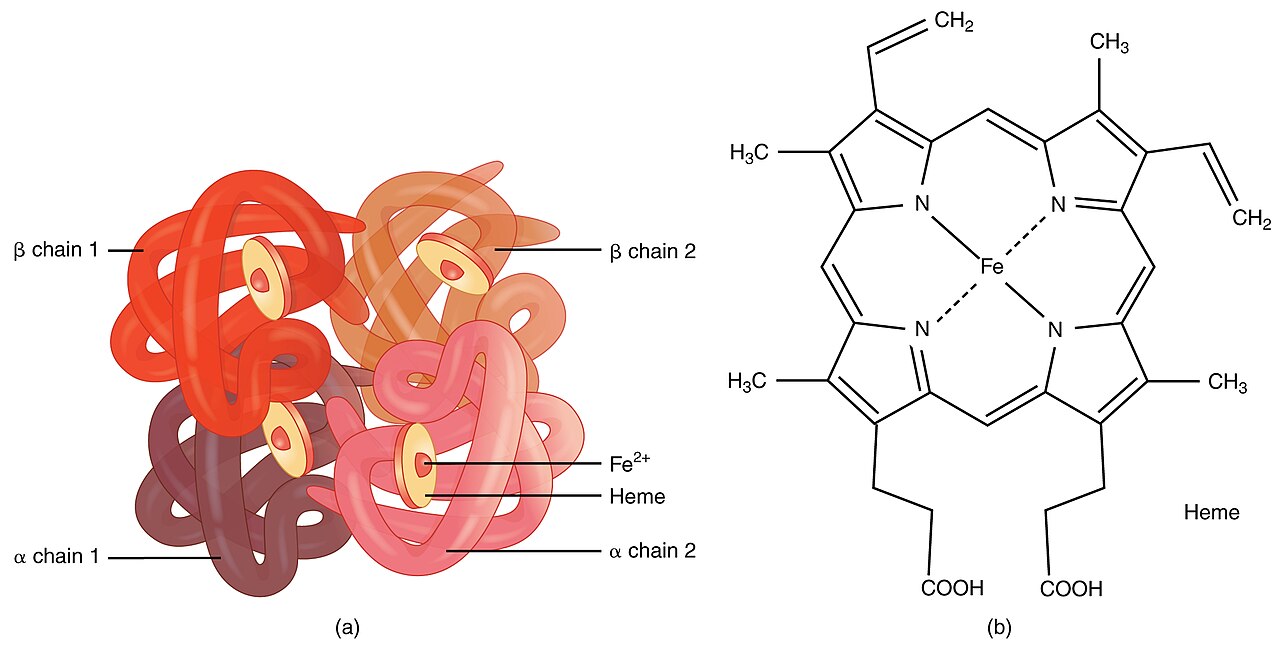


Típusok és genetika
A thalassaemia két fő csoportja létezik:
- Alfa-thalassaemia: az alfa-globin gének (általában négy példány – két HBA1 és két HBA2) érintettek; a deléciók és más mutációk a termelés csökkenését okozzák. Klinikai súlyossága a hiányzó gének számától függ (tünetmentes hordozótól súlyos fetalis formáig).
- Béta-thalassaemia: a béta-globin (HBB) gén mutációi csökkentik vagy megszüntetik a béta-globin termelését. Ennek spektruma a thalassaemia trait-től (hordozó) a thalassaemia major (Cooley-féle anémia) súlyosságáig terjed.
Öröklődés
A legtöbb thalassaemia autoszomális recesszív módon öröklődik: ha mindkét szülő hordozó (heterozigóta), az utódoknak 25% esélye van betegként (homozigóta vagy súlyos kombináció), 50% esélye hordozónak, és 25% esélye egészségesnek. Vannak azonban olyan kombinációk és mutációk, amelyek eltérő klinikai képet adhatnak.
Tünetek
A tünetek súlyossága a thalassaemia típusától és a mutációk mértékétől függ. Gyakori tünetek:
- fáradékonyság, gyengeség, sápadtság (vérszegénység)
- sárgaság (bazális hemolízis miatt)
- légszomj, csökkent terhelhetőség
- növekedési késés, késleltetett nemi éréssel járó fejlődési problémák
- megnagyobbodott lép és máj (splenomegalia, hepatomegalia)
- jellegzetes arccsonti deformitások és koponyaformálódás súlyos, hosszú ideje fennálló esetekben
Diagnózis
- alaplabor: teljes vérkép (microcytosis, alacsony hemoglobin), retikulocitaszám
- perifériás kenet: kis, hipokrom vörösvértestek, célvörösvérsejtek
- hemoglobin elektroforézis vagy HPLC: a hemoglobinfrakciók megváltozása (pl. HbA2, HbF emelkedett béta-thalassaemiában)
- molekuláris genetikai vizsgálatok: pontos mutációk kimutatása (fontos a családi tanácsadásnál és prenatális diagnosztikában)
- prenatális diagnózis: magzatvíz-vizsgálat vagy chorionboholy-mintavétel esetleg elvégezhető ismert családi mutációk esetén
Szövődmények
A betegség és a kezelések hosszú távú hatásai közé tartoznak:
- vas-túlterhelés (hemotranszfúziók és fokozott vasfelszívódás miatt) — vesekárosodás, májcirrózis, szívelégtelenség (vas-kardiomiopátia)
- endokrin zavarok: hipogonadizmus, növekedési problémák, cukorbetegség
- fertőzések fokozott kockázata (splenectomia után még inkább)
- csontdeformitások és osteoporosis
- véralvadási zavarok és trombózis bizonyos esetekben
Kezelés
A kezelés célja a tünetek csökkentése, a szövődmények megelőzése és az életminőség javítása. Fontos, hogy a terápia egyénre szabott legyen.
- Vértranszfúziók: súlyos (major) esetekben rendszeres transzfúziókra lehet szükség a normális vérszint fenntartásához.
- Vaskeláció (iron chelation): hosszú távú transzfúziók esetén elengedhetetlen a vas felhalmozódásának kezelése. Gyakori szerek: deferoxamin, deferasirox, deferiprone.
- Csontvelő- vagy őssejt-átültetés (HSCT): potenciálisan kuratív lehet, ha HLA-kompatibilis donor áll rendelkezésre; a siker függ a beteg korától, klinikai állapotától és a donor-megfelelőségtől.
- Gyógyszeres és támogató kezelés: folsavpótlás, megfelelő védőoltások (például pneumococcus), antibiotikum-profilaxis szükség szerint, növekedést és hormonális problémákat kezelő szakellátás.
- Splenectomia: egyes esetekben a lép eltávolítása csökkentheti a transzfúzióigényt, de növeli a fertőzés és a thrombosis kockázatát.
- Új és kísérleti kezelések: génterápia (lentivírus-alapú transzdukciók, CRISPR/Cas alapú beavatkozások) ígéretes lehetőségek a jövőben; klinikai vizsgálatok folynak.
Megelőzés és tanácsadás
A családi és genetikai tanácsadás kulcsfontosságú azokban a régiókban és családokban, ahol a thalassaemia gyakori. Lehetőség van hordozószűrésre, párkapcsolati tanácsadásra és prenatális diagnosztikára, hogy a szülők informált döntést hozhassanak.
Prognózis
A prognózis nagy mértékben függ a thalassaemia típusától és a terápiás lehetőségektől. A rendszeres ellátás, a transzfúziók és hatékony vaskeláció jelentősen javítják a túlélést és életminőséget. A csontvelő-átültetés gyógyulást eredményezhet alkalmas esetekben.
Epidemiológia
A thalassaemia leggyakoribb a mediterrán térségben, a Közel-Keleten, Dél-Ázsiában, Észak- és Nyugat-Afrikában, valamint Délkelet-Ázsiában. A betegség előfordulását részben a malária elleni részleges védettség magyarázza a hordozóknál, ami evolúciós előnyt adott korábban endémiás területeken.
Fontos tanácsok betegeknek és családoknak
- Tartsák rendszeresen a kapcsolatot hematológussal és gondozó centrummal.
- Következtesék a transzfúziók és vaskelációs kezelések rendjét; saját döntéseket ne hozzanak a kezelések megszakításáról.
- Vegyék igénybe a genetikai tanácsadást gyermekvállalás előtt, ha családi anamnézisben thalassaemia szerepel.
- Figyeljenek a fertőzések megelőzésére (oltások, higiénia), különösen splenectomiát követően.
Amennyiben a thalassaemia gyanúja felmerül, forduljon orvoshoz további vizsgálatokért és személyre szabott kezelésért.
Kérdések és válaszok
K: Mi az a thalassaemia?
V: A talasszaemia a vér genetikai rendellenessége, amely a mediterrán térségből származik. Ezt a betegséget a vörösvértestek gyengülése és pusztulása okozza a mutáns gének miatt, amelyek befolyásolják, hogy a szervezet hogyan állítja elő a hemoglobint.
K: Milyen szövődményekkel jár a thalassaemia?
V: A talasszaemiával kapcsolatos szövődmények közé tartozhat a tüdőgyulladás, a vastúlterhelés, a csontdeformitások és a szív- és érrendszeri betegségek.
K: Hogyan nyújt védelmet a thalassaemia a malária ellen?
V: A thalassaemia hordozói szelektív túlélési előnyt élveznek (úgynevezett heterozigóta előny), ami segít a mutáció mutációs rátáját jóval meghaladó populációkban tartani. Ez védelmet nyújt számukra a malária ellen, amely gyakori vagy gyakori volt azokban a régiókban, ahol ez a tulajdonság gyakori.
K: Vannak-e a thalassaemiának különböző változatai?
V: Igen, a thalassaemiának számos különböző változata létezik, amelyek mindegyike a genom különböző pozíciójában bekövetkező mutáció miatt alakul ki. Hasonlít egy másik, a hemoglobint érintő genetikai rendellenességhez, a sarlósejtes betegséghez.
K: Lehetséges-e gyógyítani a thalassaemiás betegeket?
V: Igen, lehetséges a talasszémiás betegek gyógyítása olyan kompatibilis donorokból származó csontvelőátültetéssel, akiknek van HLA-egyeztetett kompatibilis donorjuk.
Kapcsolódó cikkek
Szerző
AlegsaOnline.com Thalassaemia – meghatározás, okai, tünetei és kezelése Leandro Alegsa
URL: https://hu.alegsaonline.com/art/97356
Források
- accessmedicine.com : accessmedicine.com/content.aspx?aID=6123722
- mayoclinic.com : mayoclinic.com/health/thalassemia/DS00905/DSECTION=complications
- bloodjournal.hematologylibrary.org : HLA-matched sibling bone marrow transplantation for β-thalassemia major